Point de pratique
Les anomalies vasculaires pendant l’enfance : quand traiter les patients et quand les diriger vers une ressource spécialisée

Affichage : le 5 août 2022
Auteur(s) principal(aux)
Kelley Zwicker, Julie Powell, Carl Cummings; Société canadienne de pédiatrie, Comité de la pédiatrie communautaire
Paediatr Child Health 2022 27(5):315–319. 
Résumé
Les anomalies vasculaires sont des affections hétérogènes qui touchent les vaisseaux sanguins ou lymphatiques. Les enfants atteints peuvent éprouver de la douleur ou une perte fonctionnelle, présenter une infection ou une coagulopathie ou être confrontés à des difficultés psychologiques. Le diagnostic et la prise en charge exigent souvent une approche interdisciplinaire. Sept cliniques d’anomalies vasculaires au Canada offrent des soins interdisciplinaires. Le présent point de pratique propose une approche thérapeutique des anomalies vasculaires pédiatriques les plus fréquentes (hémangiomes). On y passe en revue les indications de diriger les patients vers une clinique spécialisée, en s’attardant sur les anomalies vasculaires complexes, et notamment les hémangiomes infantiles, qui peuvent provoquer des complications.
Mots-clés : anomalie vasculaire; hémangiome congénital; hémangiome infantile; propranolol
Historique et prévalence
Le système de classification des anomalies vasculaires est établi par l’International Society for the Study of Vascular Anomalies (ISSVA). Ces anomalies se divisent en deux groupes : les tumeurs (qui sont prolifératives) et les malformations (figure 1) [1], qui sont définies selon le type de vaisseau anormal (capillaire, artériel, veineux ou lymphatique). Les termes historiques, comme « tache framboisée » et « tache de vin », sont déconseillés (tableau 1). La nomenclature de l’ISSVA favorise une meilleure communication entre les spécialistes qui soignent les patients présentant ces anomalies.
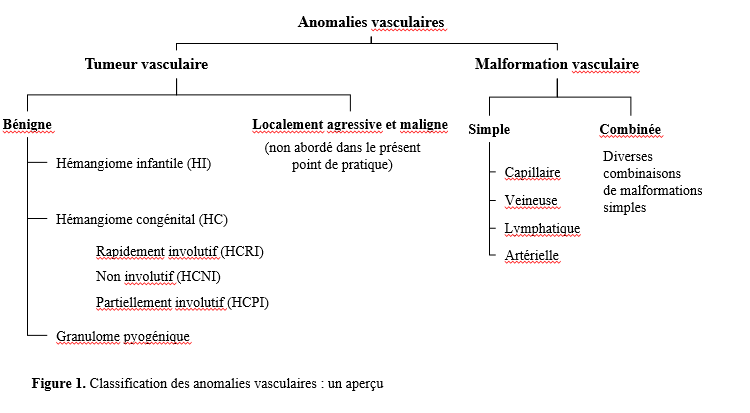
On estime que les hémangiomes touchent de 4 % à 10 % des nourrissons [2]. Puisqu’ils ont tendance à involuer (diminuer de volume) spontanément, on suppose souvent qu’ils se résorbent sans complication. Pourtant, jusqu’à 15 % des hémangiomes peuvent s’accompagner de complications, qui prennent surtout la forme d’ulcération, de douleur, d’atteinte fonctionnelle permanente ou d’aspect esthétique insatisfaisant [3]. Les professionnels de la santé doivent connaître les types d’hémangiomes et leur évolution naturelle pour donner des conseils, contribuer à orienter les attentes des patients ou des parents et traiter ces anomalies de manière appropriée. Les hémangiomes infantiles (HI) et congénitaux (HC) sont au cœur du présent point de pratique.
Les hémangiomes infantiles
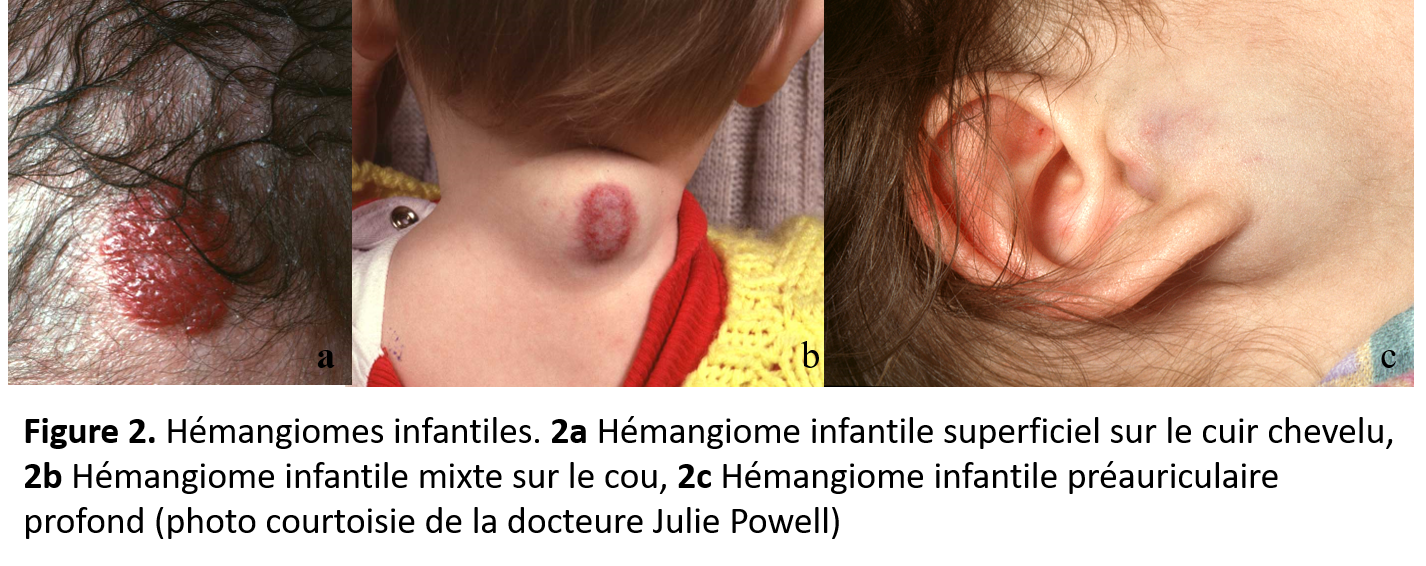
Les HI (figure 2) sont beaucoup plus fréquents que les HC (figure 3). Ils présentent une phase caractéristique de croissance et de régression. Ainsi, la phase proliférative s’amorce chez le nourrisson et devient souvent perceptible dans les quatre premières semaines de vie [4]. Elle se caractérise par une croissance rapide de un à trois mois après la naissance. La plupart des hémangiomes continuent de croître entre cinq et 12 mois de vie, et cette croissance est suivie d’un plateau, généralement entre 12 et 18 mois de vie. La phase d’involution se produit entre l’âge de un à sept ans, mais selon les récentes études, le processus d’involution de 90 % des HI est terminé à l’âge de quatre ans. Du tissu fibroadipeux résiduel, une atrophie, des cicatrices ou de la peau superflue, accompagnés ou non de télangiectasies, persistent dans environ 50 % des cas d’HI non traités [5][6].
Par le passé, les HI compliqués étaient traités à l’aide de stéroïdes systémiques [2], mais cette approche n’a plus cours en raison de leurs effets secondaires et de la découverte, en 2008, de l’efficacité et du meilleur profil d’innocuité des bêtabloquants oraux, qui sont désormais considérés comme le traitement de première intention. Le traitement au laser est possible, mais cette intervention est généralement réservée aux hémangiomes ulcérés qui ne répondent pas aux bêtabloquants, ou aux situations où des télangiectasies persistent après l’involution. Il n’y a pas de lignes directrices consensuelles canadiennes au sujet du traitement de l’HI, même si l’utilisation de bêtabloquants par voie orale est une pratique bien établie dans le monde pour traiter l’HI compliqué ou à risque de complications. Les indications d’amorcer l’administration de propranolol incluent les HI qui représentent une menace fonctionnelle ou vitale comme l’atteinte des voies respiratoires, les HI périoculaires, l’emplacement sur la lèvre, sur le nez (notamment la pointe nasale) ou dans le conduit auditif, l’ulcération, les hémangiomes segmentaires du visage ou les HI comportant un risque de défiguration permanente [2][7][8].
Au Canada, Santé Canada a approuvé une formulation de propranolol du nom d’Hemangiol. Deux lignes directrices publiées recommandent de commencer par administrer 1 mg/kg/jour de propranolol, divisé en deux prises [2][7]. La dose est ensuite accrue de 0,5 mg/kg/jour à 1 mg/kg/jour toutes les semaines, jusqu’à concurrence de 2 mg/kg/jour à 3 mg/kg/jour, toujours répartie en deux prises [9]. Il est proposé d’accroître la dose plus lentement chez les nouveau-nés et les nourrissons prématurés de moins de deux mois d’âge corrigé ou en cas de soupçon de syndrome PHACE (désignant un grand HI segmentaire sur le visage ou le cou, associé à l’une ou l’autre des anomalies suivantes : fosse postérieure, hémangiome, anomalies artérielles cérébrales ou cervicales, coarctation de l’aorte ou autres malformations cardiaques et anomalies oculaires – qui correspond à eyes en anglais) [10]. Il est recommandé d’ajuster la dose en fonction du poids.
Avant de commencer à administrer du propranolol, il est essentiel de vérifier le rythme cardiaque, la présence de souffle cardiaque ou de bruits cardiaques anormaux et les pouls périphériques, et il faut aussi envisager un électrocardiogramme et un échocardiogramme lorsque l’examen cardiaque est anormal. Dans certains centres, la fréquence cardiaque et la tension artérielle sont mesurées sur une période de une à deux heures après la première dose, puis après chaque augmentation de la dose. Cette pratique varie selon les centres. Il est recommandé d’entreprendre l’administration de propranolol en milieu hospitalier chez les nourrissons ayant des troubles cardiorespiratoires connexes, chez les nourrissons de moins de cinq semaines de vie et chez les enfants de familles dont le soutien social est sous-optimal [2]. Le suivi habituel des patients dont la dose est stable et qui ne présentent pas de complications a généralement lieu tous les deux ou trois mois, mais dépend également de chaque centre.
Les perturbations du sommeil et la froideur ou les marbrures des mains et des pieds sont des effets secondaires courants du traitement au propranolol. L’hypotension, le bronchospasme, l’hypoglycémie, la bradycardie et l’hyperkaliémie font partie des effets secondaires moins courants. L’hypoglycémie, le bloc cardiaque du deuxième ou troisième degré et l’hypersensibilité au propranolol sont des contre-indications absolues à ce médicament [2][7][8]. L’hypoglycémie est une complication potentielle grave. Il est recommandé de conseiller aux parents d’administrer la dose aux alentours de l’heure des boires, le matin et le soir, et de rester à l’affût des symptômes d’hypoglycémie (p. ex., sudation, tachycardie, aspect d’anxiété ou d’agitation). On a observé des cas d’hypoglycémie spontanée après des changements de dose marqués ou particulièrement lorsque le propranolol continue d’être administré malgré une maladie. Il est recommandé d’interrompre le propranolol en cas de maladie, en raison des risques d’apport oral réduit et de déshydratation.
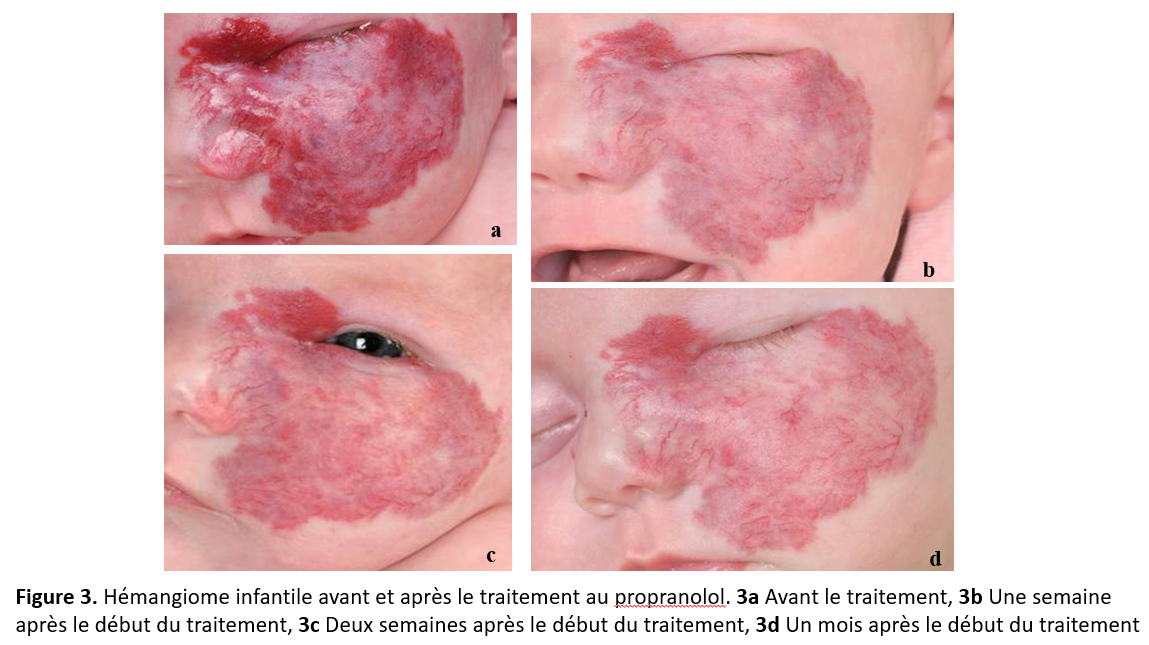
En général, le traitement au propranolol se poursuit pendant au moins six mois, mais la réponse initiale est habituellement visible dans les trois à quatre semaines suivant le début du traitement. Un changement de coloration est souvent observé dans les 48 premières heures de traitement (figure 3). L’involution de la lésion ou l’absence de réponse constituent des indications pour mettre un terme au traitement. Les parents doivent être informés que, dans certains cas, un rebond de la croissance peut se produire après l’arrêt de la médication [11]. Ils devraient également être avisés que l’hémangiome risque de ne pas se résorber complètement et que du tissu fibroadipeux résiduel, une atrophie ou des télangiectasies peuvent persister.
Un certain nombre d’essais cliniques a permis d’évaluer le timolol topique pour traiter l’HI [12]. Ce médicament, prescrit sous forme de solution gélifiante ophtalmique de maléate de timolol à 0,5 %, est une possibilité en cas de HI petits et superficiels. Une ou deux gouttes sont administrées deux fois par jour, quelle que soit la taille de l’hémangiome. La pharmacocinétique du timolol est mal comprise, et on ne sait pas à quel moment les taux sériques de pointe sont atteints après une application cutanée. Il ne faut pas prescrire plus de gouttes en cas de lésions plus grosses. De plus, l’application topique sur un hémangiome ulcéré n’est pas nécessairement sécuritaire en raison d’une plus grande absorption. En cas d’HI grand ou ulcéré, le traitement au propranolol par voie orale est préférable au timolol par voie cutanée.
Les hémangiomes congénitaux
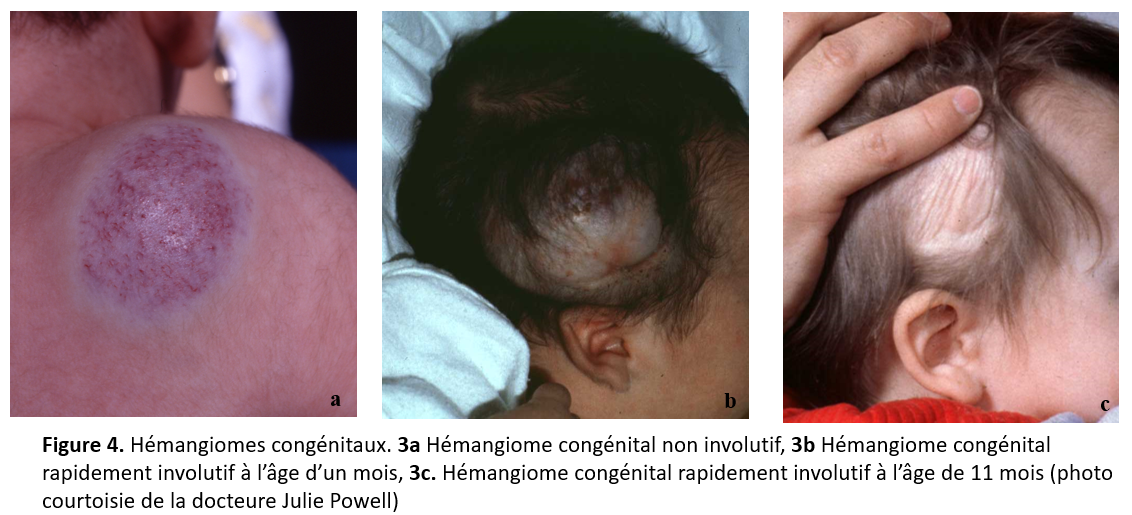
Les HC sont différents des HI (figure 4) parce qu’ils sont déjà pleinement développés à la naissance [1]. L’HC doit être distingué de l’HI, qui est parfois présent à la naissance sous forme de tache vasculaire, de télangiectasie ou de lésion ecchymotique, mais qui, le plus souvent, croît entre les quelques premières semaines et les quelques premiers mois de vie. À la naissance, les HC prennent la forme de plaques ou de tumeurs vasculaires sans prolifération précoce [13]. Les trois sous-types majeurs d’HC sont l’HC rapidement involutif, l’HC non involutif et l’HC partiellement involutif [14] (figure 4). L’HC ne répond pas aux stéroïdes ni aux bêtabloquants, et les données probantes à jour n’appuient pas l’utilisation des bêtabloquants pour traiter plus particulièrement les HC non involutifs et partiellement involutifs. Il n’est généralement pas indiqué de traiter les HC rapidement involutifs, justement en raison de leur involution spontanée rapide. Lorsque les HC s’ulcèrent ou constituent un risque esthétique ou fonctionnel, il est bon de diriger le nourrisson vers une clinique d’anomalies vasculaires ou en dermatologie pédiatrique.
Quand traiter les patients et quand les diriger vers une ressource spécialisée
Les petits hémangiomes non ulcérés qui ne sont pas situés à un endroit esthétiquement problématique peuvent souvent être soignés par le médecin de première ligne. Il est recommandé de diriger le patient vers une ressource spécialisée en présence d’un hémangiome qui touche l’œil, le nez, l’urètre, l’anus ou le foie ou d’un grand hémangiome du visage. En présence de plus de cinq hémangiomes cutanés, il faut envisager une échographie abdominale afin d’écarter la possibilité d’hémangiomatose hépatique connexe. Tous les grands hémangiomes infantiles segmentaires du visage sont toutefois à évaluer, au cas où ils seraient associés à des troubles du syndrome PHACE. Un traitement précoce contribue à assurer une issue optimale. Ainsi, les cas plus complexes doivent être dirigés le plus rapidement possible vers une ressource spécialisée, de préférence avant l’âge de quatre mois.
Des ulcérations se forment dans environ 15 % des cas d’HI, particulièrement pendant la phase de croissance rapide. Il est difficile de prédire la durée de la guérison. Comme la douleur et les infections sont de fréquentes complications qui doivent être traitées rapidement, il est conseillé de diriger les patients vers une ressource spécialisée. Lorsque l’hémangiome empiète sur une structure essentielle ou lui nuit, il est préférable d’opter pour un traitement plutôt que pour une « prise en charge non interventionniste ». Il faut également diriger vers une ressource spécialisée les patients qui éprouvent des effets indésirables au traitement au propranolol ou qui n’y réagissent pas. Dans de telles situations, d’autres bêtabloquants par voie orale (nadolol ou aténolol), des stéroïdes par voie orale ou, dans de rares cas, une résection chirurgicale pourraient s’imposer. Les traitements ne sont pas toujours curatifs, mais ils peuvent atténuer des effets d’ordre fonctionnel et esthétique.
D’autres anomalies vasculaires sont caractérisées dans la classification de l’ISSVA et doivent être distinguées des HI. Les malformations capillaires prennent la forme de taches planes congénitales, rosées à rouges [1], qui peuvent se manifester n’importe où sur le visage ou le corps. Les malformations veineuses désignent des veines anormales comportant une couche de cellules musculaires lisses défectueuse. Le sang y circule lentement et a tendance à former des phlébolithes, ce qui peut créer une certaine douleur. Les malformations artérioveineuses sont des connexions anormales entre les artères et les veines, dénuées de lits capillaires. Quant aux malformations lymphatiques, elles sont caractérisées par leur taille (microkystiques ou macrokystiques), dont l’anomalie macrokystique du cou représente une variante. Les syndromes d’hypercroissance (p. ex., spectre d’hypercroissance lié au gène PIK3CA) sont également associés à des anomalies vasculaires. Ces cas plus complexes doivent être dirigés vers une clinique d’anomalies vasculaires. Les possibilités actuelles de traitement des malformations vasculaires sont variées et incluent des mesures conservatrices (soulagement de la douleur, vêtements compressifs), des interventions (sclérothérapie, intervention chirurgicale) ou une approche combinée. Les médicaments ciblés qui ont récemment été mis en marché pour traiter des malformations vasculaires complexes (p. ex., sirolimus) sont prometteurs, mais dépassent la portée du présent point de pratique.
Principaux points de pratiques au sujet des hémangiomes infantiles et congénitaux chez les enfants
- Les soins essentiels des hémangiomes incluent un examen physique et dermatologique complet et la prise en considération du diagnostic différentiel. Il est préconisé d’aborder les préoccupations psychosociales des patients et des parents de manière proactive.
- Les hémangiomes n’ont pas tous besoin d’être traités. Les indications d’entreprendre le traitement incluent le risque d’atteinte fonctionnelle ou vitale, l’emplacement, les répercussions esthétiques et l’ulcération.
- Un traitement précoce s’impose pour obtenir un résultat optimal, et c’est pourquoi il est essentiel de diriger le patient le plus rapidement possible vers une ressource spécialisée, idéalement avant l’âge de quatre mois.
- L’utilisation d’une terminologie appropriée facilite la communication avec les parents d’enfants touchés et entre les spécialistes qui participent à la prise en charge des hémangiomes pédiatriques.
Remerciements
Les auteurs souhaitent remercier les docteures Elena Pope (chef d’unité, dermatologie pédiatrique, SickKids) et Miriam Weinstein (médecin, dermatologie pédiatrique, SickKids) d’avoir révisé le présent point de pratique, qui a également été révisé par le comité de la pharmacologie et des substances dangereuses de la Société canadienne de pédiatrie.
COMITÉ DE LA PÉDIATRIE COMMUNAUTAIRE DE LA SOCIÉTÉ CANADIENNE DE PÉDIATRIE (2019-2020)
Membres : Julia Orkin MD (présidente), Marianne McKenna MD (représentante du conseil), Tara Chobotuk MD, Michael Hill MD, Audrey Lafontaine MD, Alisa Lipson MD, Carl Cummings MD (président sortant), Larry Pancer MD (membre sortant)
Représentant : Peter Wong (représentant de la section de la pédiatrie communautaire)
Auteurs principaux : Kelley Zwicker MD, Julie Powell MD, Carl Cummings MD
Références
- Mulliken JB, Burrows PE, Fishman SJ, éd. Mulliken and Young’s Vascular Anomalies: Hemangiomas and Malformations. New York, NY: Oxford University Press; 2013.
- Krowchuk DP, Frieden IJ, Mancini AJ et coll. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics 2019;143(1):e20183475.
- Chamlin SL, Haggstrom AN, Drolet BA et coll. Multicentre prospective study of ulcerated hemangiomas. J Pediatr 2007;151(7):684-89,689.e1.
- Tollefson MM, Frieden IJ. Early growth of infantile hemangiomas: What parent’s photographs tell us. Pediatrics 2012;130(2):e314-20.
- Chen TS, Eichenfield LF, Friedlander SF. Infantile hemangiomas: An update on pathogenesis and therapy. Pediatrics 2013;131(1):99–108.
- Liang MG, Frieden IJ. Infantile and congenital hemangiomas. Semin Pediatr Surg 2014;23(4):162–7.
- Solman LM, Glover M, Beattie PE et coll. Oral propranolol in the treatment of proliferating infantile haemangiomas: British Society for Paediatric Dermatology consensus guidelines. Br J Dermatol 2018;179(3):582–9.
- Martin K, Blei F, Chamlin SL et coll. Propranolol treatment of infantile hemangiomas: Anticipatory guidance for parents and caretakers. Pediatr Dermatol 2013;30(1):155–9.
- Léauté-Labrèze C, Hoeger P, Mazereeuw-Hautier J et coll. A randomized, controlled trial of oral propranolol in infantile hemangioma. N Engl J Med 2015;372(8):735-46.
- Garzon MC, Epstein LG, Heyer GL et coll. PHACE syndrome: Consensus-derived diagnosis and care recommendations. J Pediatr 2016;178:24-33.e2.
- Shah SD, Baselga E, McCuaig C et coll. Rebound growth of infantile hemangiomas after propranolol therapy. Pediatrics 2016;137(4):e20151754.
- Püttgen K, Lucky A, Adams D et coll. Topical timolol maleate treatment of infantile hemangiomas. Pediatrics 2016;138(3): e20160355.
- North PE, Waner M, James CA, Mizeracki A, Frieden IJ, Mihm MC. Congenital nonprogressive hemangioma: A distinct clinicopathologic entity unlike infantile hemangioma. Arch Dermatol 2001;137(12):1607-20.
- Nasseri E, Piram M, McCuaig CC, Kokta V, Dubois J, Powell J. Partially involuting congenital hemangiomas: A report of 8 cases and review of the literature. J Am Acad Dermatol 2014;70(1):75–9.
Avertissement : Les recommandations du présent document de principes ne constituent pas une démarche ou un mode de traitement exclusif. Des variations tenant compte de la situation du patient peuvent se révéler pertinentes. Les adresses Internet sont à jour au moment de la publication.
Mise à jour : le 8 février 2024